November 22, 2018
Overflowing with antibodies and optimism
There is more optimism than ever that researchers are on the path to developing vaccines and antibodies that can help stem HIV’s persistent spread.
Kristen Jill Kresge
“A new era. An exciting time. More optimism than ever before.” These are just some of the ways researchers are describing the current state of play in HIV vaccine and antibody research.
“There is very exciting new research that gives us great hope that we are making substantial progress with vaccines and antibodies,” says John Mascola, director of the Vaccine Research Center (VRC) at the U.S. National Institute of Allergy and Infectious Diseases. “We have better vaccine antigens in the last few years than we’ve ever had, and also better vaccine platforms. An effective vaccine is likely, and we’re on that path.”
This overarching sense of optimism was palpable when thousands of researchers gathered in Madrid, Spain, this October for the biennial HIV Research for Prevention (HIVR4P) conference. With two vaccine candidates in ongoing efficacy trials, numerous other candidates entering clinical testing, and a flurry of activity in developing antibodies for prophylaxis, the field seems undeniably enthusiastic about the prospects for developing new HIV prevention strategies.
Since 2009, when the first of what now amounts to hundreds of potent antibodies with the ability to broadly neutralize HIV were isolated from HIV-infected volunteers, researchers have gained a remarkable understanding of just how these antibodies form in response to natural HIV infection.
HIV is coated with bulky sugar molecules that themselves are not immunogenic and largely deflect immune responses mounted against the virus. “Antibodies prefer to see proteins,” says Mascola. “It’s very hard for an antibody to navigate that glycan shield, find a protein, and neutralize.” Yet some HIV-infected individuals can and do make the types of broad and potent antibodies known as broadly neutralizing antibodies (bNAbs) a vaccine would ideally induce. What researchers have found is that despite its glycan shield, HIV’s outer protein known as Envelope (Env) actually has many sites of vulnerability. “Virtually the entire Env can be targeted [by antibodies],” says Andrew Ward, a professor at Scripps Research in La Jolla, CA, and a principal investigator of IAVI’s Neutralizing Antibody Center (NAC).
This doesn’t, however, mean that bNAbs form easily. There are a myriad of reasons why bNAbs are unusual and don’t develop often or quickly in response to natural infection. “Most B cells do not have the intrinsic capacity to make broadly neutralizing antibodies,” says Rogier Sanders, adjunct associate professor of research in microbiology and immunology at Weill Cornell Medicine and a professor of virology at the University of Amsterdam.
Another reason is that many protective antibody lineages are disfavored by the immune system and are therefore controlled by immune tolerance mechanisms. In a recent study comparing nearly 50 HIV-infected individuals whose immune systems generate bNAbs with a similar number of individuals who did not, researchers found evidence of natural killer (NK) cell dysfunction in the individuals who made bNAbs (Cell 175, 387, 2018). This NK cell dysfunction resulted in higher levels of follicular T-helper (Tfh) cells or dendritic cells that support B-cell responses in those individuals whose immune systems make bNAbs compared to those who don’t. This finding suggests that the ability to induce bNAbs through vaccination may be aided by modulating NK cell activity.
At HIVR4P, assistant professor of medicine Todd Bradley from Duke University, who led this study, reported on his lab’s efforts to study how modulating immune tolerance mechanisms might affect the development of neutralizing antibody responses following vaccination. They found that in rhesus macaques, depletion of NK cells by the cytokine interleukin (IL)-15 enhanced formation of germinal centers and Tfh responses following immunization, and that this resulted in higher levels of both binding and neutralizing antibodies in the IL-15 treated animals as compared to controls.
Other studies were designed to see if transient modulation of the immune checkpoint molecules CTLA-4 and PD-1 would in any way alter antibody responses that develop following immunization with HIV Env in macaques. This year’s Nobel Prize in Physiology or Medicine was awarded to two scientists for their discovery of cancer therapies based on inhibition of these immune checkpoint molecules that function as brakes on the immune system. Bradley and colleagues found that while inhibiting CTLA-4 augmented the development of antibody responses to HIV Env with the ability to neutralize the less challenging tier-1 viruses, inhibiting PD-1 actually resulted in lower levels of antibodies that could bind to or neutralize HIV when co-administered with an HIV vaccine.
They also tested whether inhibiting CTLA-4 or the co-stimulatory immune checkpoint molecule OX-40 would have any effect on antibody responses following a prime-boost vaccination with the engineered germline-targeting vaccine immunogen known as eOD-GT8 60mer and the native-like trimeric protein known as BG505 SOSIP in a humanized VRC01 bNAb precursor knock-in mouse model. They found that blocking CTLA-4 and OX-40 agonism increased the antibody titers in the vaccinated mice. The next step is to determine whether depleting NK cells along with inhibiting CTLA-4 and stimulating OX-40 would have a synergistic effect. While still in early stages, efforts to understand how immune system modulation might encourage the formation of bNAbs is an active area of study. This work led William Schief, a professor in the department of immunology and microbiology at Scripps Research and a principal investigator in IAVI’s NAC, to conclude that “immune system modulation has legs.”
Another reason that bNAbs are unique is that they are almost always heavily mutated, or matured, as a result of undergoing repeated rounds of somatic hypermutation in germinal centers in response to an evolving and ever-mutating virus. This is why in most cases it takes between three to five years of chronic stimulation before the human immune system can generate potent and broadly cross-reactive antibodies against HIV.
This presents a challenge for vaccine researchers. While they want to develop a vaccine that can mimic the process of bNAb development in natural HIV infection, “we don’t want to recapitulate the timeframe it takes,” says Kevin Wiehe, assistant professor in medicine at Duke University. “What we’re looking for are shortcuts.”
Wiehe and colleagues have found that broadly reactive neutralizing antibodies are more likely to have what they call improbable genetic mutations, but that not all of these mutations are critical. Wiehe and others are trying to identify the individual or subset of improbable mutations that can have the biggest effect on bNAb development, and then are focusing on ways to induce only these mutations in an effort to speed up the maturation process of antibodies. “These critical and improbable mutations are what we need to go after with vaccine immunogens,” says Wiehe. This mutation-guided approach to vaccine design is just one method researchers are exploring to induce mature bNAbs as quickly as possible through vaccination.
After decades of largely disappointing results from clinical trials, the HIV vaccine field has entered a new phase. There are two ongoing vaccine efficacy trials, one of which is testing a reformulated version of the only vaccine regimen to date that offered any protection against HIV infection in the RV144 efficacy trial (HVTN 702; see Awaiting Results from Efficacy Trials, IAVI Report, Vol. 22, No. 2, 2018). The other is testing a vaccine regimen based on an adenovirus serotype 26 (Ad26) mosaic candidate-based regimen that provided promising results in both preclinical animal studies and early phase clinical trials in humans (HVTN 705; see The Imbokodo Phase IIb HIV vaccine trial).
Now there are even tantalizing clues that suggest these two vaccine candidates may be activating similar genetic pathways. At HIVR4P, Rasmi Thomas, chief of the host genomics section at the U.S. Military HIV Research Program, reported that a vaccine-induced B-cell pathway that was found to be associated with partial protection against HIV in RV144 vaccine recipients was also detected in non-human primates that were protected against simian immunodeficiency virus (SIV) in two preclinical studies of the Ad26 mosaic-based regimen. The enriched genes in this pathway are involved in B-cell development and proliferation, as well as toll-like receptor signaling, and are associated with a higher magnitude of antibody-dependent cellular phagocytosis, according to Thomas. Though she warns that “a lot of this is preliminary.” Previous studies have also shown that this genetic pathway was associated with higher antibody responses to both influenza and yellow fever vaccination.
In addition to these efficacy trials, there are also several new vaccine constructs either in or about to enter Phase I clinical trials. Many of these candidates are based on designer immunogens that aim to induce long sought-after bNAb responses against the virus. Hundreds of bNAbs have been identified and some of them are effective at protecting against an SIV/HIV hybrid in non-human primate studies. “The coupling of these bNAbs and the trimer with structural studies have immensely facilitated structure-guided immunogen design and have made the development of an HIV neutralizing antibody vaccine appear to be an achievable goal,” according to a review article by Raiees Andrabi and colleagues at Scripps Research and IAVI’s NAC (Curr. Opin. Immunol. 53, 143, 2018).
The new crop of vaccine candidates now entering clinical trials hinges primarily on four major approaches to inducing bNAbs, all of which are complementary and in many cases are being studied in combination. The first is referred to as lineage-based vaccine design. This approach is based on detailed study of the co-evolution of HIV and antibodies in infected individuals in real time and attempts to mimic this antibody maturation process with a series of immunogens that are derived from sequential HIV Env variants.
One of the lineage-based approaches in development by Barton Haynes, director of the Human Vaccine Institute at the Duke University School of Medicine, and colleagues at the Duke Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery (CHAVI-ID) is already in clinical trials (HVTN 115). This trial is testing a series of immunogens based on viruses identified from a single individual, referred to as CH505, who was enrolled in an acute infection study and followed from the time HIV infection occurred through the development of bNAb responses. In the ongoing Phase I HVTN 115 trial, researchers are testing a series of sequential HIV gp120 immunogens administered along with a GLA-SE adjuvant either alone or in combination with a DNA mosaic-based candidate. A trial similar to HVTN 115 is also being proposed in infants, given that the immune systems of infants and children may more readily generate bNAbs. Many other lineage-based approaches are also in development (see table, above).
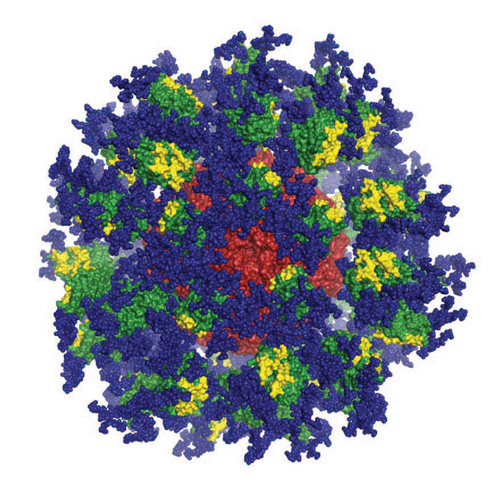 An image of the eOD-GT8 60mer engineered immunogen. Image courtesy of Scripps Research/IAVI’s Neautralizing Antibody Center.
An image of the eOD-GT8 60mer engineered immunogen. Image courtesy of Scripps Research/IAVI’s Neautralizing Antibody Center.
The second major approach to inducing bNAbs through vaccination is referred to as germline-targeted vaccine design. This approach utilizes a priming immunogen—engineered based on a specific epitope on HIV that is targeted by bNAbs—to activate B cells that have the intrinsic capability to make bNAbs. This is followed by boosting with one or more HIV Env immunogens that are increasingly similar to the native structure of HIV Env to shepherd the germline antibodies to accrue the mutations that will make them broadly neutralizing. The germline-targeting candidate eOD-GT8 60mer recently entered a Phase I clinical trial (G001). This candidate was engineered by scientists at Scripps Research and IAVI’s NAC to activate B cells with the capacity to make CD4 binding-site directed antibodies.
Another germline-targeting immunogen developed by Haynes and colleagues may enter clinical trials next year. This immunogen is a stabilized trimeric version of the transmitted founder virus from the CH505 donor.
The third vaccine design approach is referred to as either an epitope-focused strategy or immunofocusing. Two candidates based on this approach will also enter clinical trials next year.
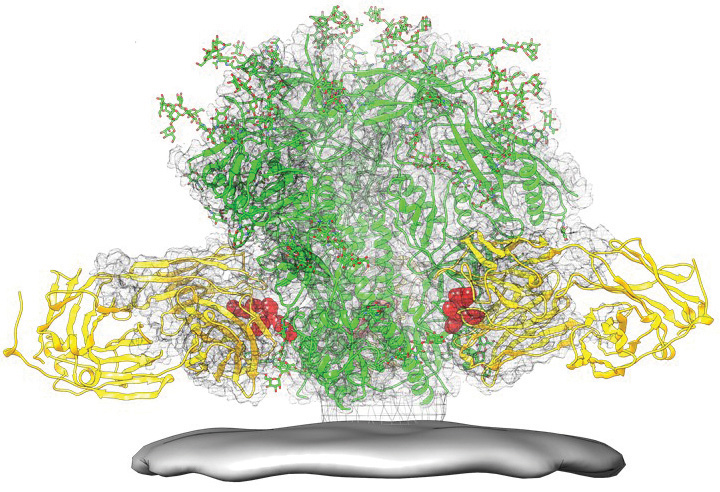
Peter Kwong, a senior investigator in the structural biology section of the VRC, and colleagues first identified an antibody referred to as VRC34 that targets the fusion peptide (FP) region of HIV Env, which plays a critical role in viral entry into cells (see image at left). Kwong and colleagues then developed an immunogen by linking this FP, which consists of eight amino acids, to a carrier protein called KLH. This immunogen was tested extensively in preclinical studies in combination with a trimeric HIV Env boost. One antibody that developed following vaccination of rhesus macaques neutralizes 59 percent of a 208 virus panel of global isolates. Now the team is modifying the immunogen by affixing it to a nanoparticle, which will make the immunogen more closely resemble a virus in size and structure, and is preparing to advance this prime-boost strategy into Phase I clinical trials next year.
Another epitope-based immunogen that is scheduled to enter clinical trials next year is based on the membrane proximal external region (MPER) epitope associated with lipid. These MPER peptide-liposomes are being developed by Haynes and colleagues at Duke.
The fourth approach is testing native-like trimers as immunogens. It is only since 2013 that researchers have been able to make stable proteins that look like the native HIV Env trimer. Doing so allowed scientists to develop a stable crystal structure of the virus for the first time and is, in addition to the isolation of bNAbs from HIV-infected volunteers, the other breakthrough that is fueling today’s vaccine design efforts.
There are now several native-like trimers in development, with the first—the BG505 SOSIP trimer developed by Sanders and John Moore of Weill Cornell Medicine—entering clinical trials imminently. Others, including those that are based on consensus sequences, are slated to enter clinical trials next year, and a modified BG505 trimer that engages germline precursors of bNAbs known as BG505 GT1.1 will also enter clinical trials in 2019.
There is also an effort underway by the European AIDS Vaccine Initiative (EAVI2020) to decipher just what makes a cohort of acutely HIV-infected individuals able to develop bNAbs much earlier in the course of infection. “They are very rare, much rarer than in chronic infection, but there are some individuals who have some level of [antibody] breadth even as soon as two to three months after infection,” says Sanders. If there is something unique about the Envs of the infecting viruses in these individuals that is driving the antibody response to develop much more rapidly, they would make ideal vaccine candidates. This is why researchers are making HIV Env proteins similar to those isolated from these individuals and will test them in clinical trials beginning next year.
“There is a huge explosion of candidates going into clinical trials,” says Haynes. “It’s going to be an exciting time.”
What’s notable, in addition to the number of candidates being investigated, is the pace at which this research is moving. “This is all three or four years old,” says Mascola. “It is really moving fast.”
At the same time, researchers are still mining for additional antibodies from human volunteers. At HIVR4P, Mohammed Sajadi, associate professor at the University of Maryland School of Medicine, reported on the identification of a single donor whose serum could neutralize 99 to 100 percent of a global panel of HIV isolates. The individual was HIV infected for almost 20 years at the time the serum samples were collected. From this serum, researchers were able to isolate three new monoclonal antibodies, dubbed N49 P6, P7, and P11, which Sajadi referred to as the “most broad and potent in vitro antibodies identified to date.”
But even with multiple strategies and budding optimism, it isn’t likely to be easy to induce bNAbs through vaccination. “There are a lot of factors we have to get right all at the same time to get this system to work,” says Haynes. “We’ve only just begun to learn the rules of how to do this.” Scientists will need to optimize the immunogens, adjuvants, and delivery systems, and potentially also explore strategies to overcome host tolerance responses. One thing almost all researchers agree on is that developing vaccines that can induce bNAbs will require iterative cycles of clinical evaluation and optimization.
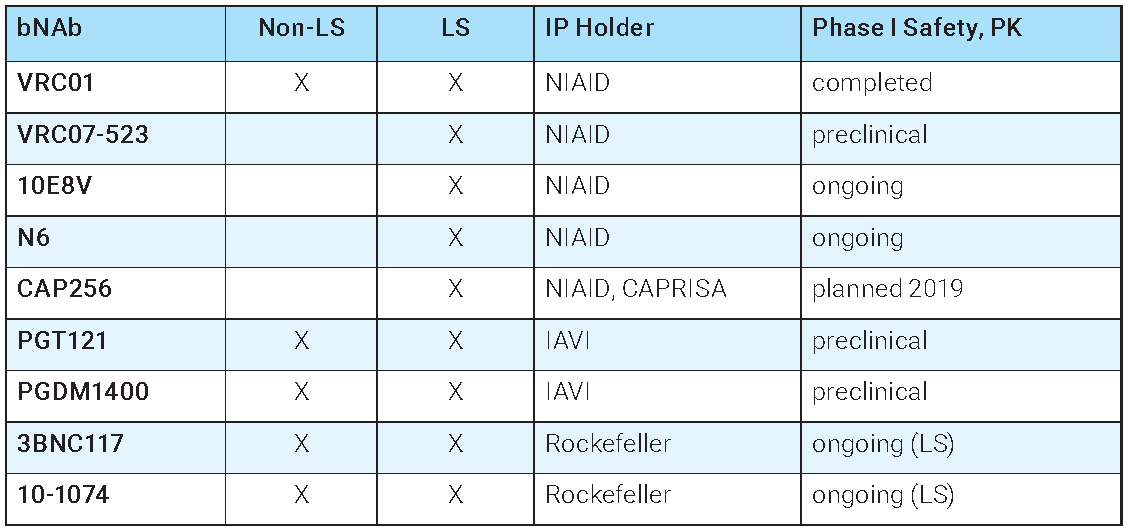

In the meantime, nine bNAbs are in clinical development for antibody-based prophylaxis—the direct administration of antibodies to prevent HIV infection. Rick Koup, a senior investigator at the VRC, says that while researchers are exploring “elaborate strategies to make bNAbs in people, the easier way is to make bNAbs in a bioreactor.”
So that is exactly what researchers are doing, and within a couple of years they will have the first human data on this approach from the Phase IIb proof of concept trials known as the Antibody-Mediated Prophylaxis or AMP studies. These studies (also known as HVTN 703 and HVTN 704) are testing whether administration of the bNAb known as VRC01 can protect against HIV infection in trials involving more than 4,600 women, men who have sex with men, and transgender individuals from multiple countries in North and South America, Europe, and Africa. Enrollment in these studies is now complete.
There is also a trial testing whether VRC01 or the variant VRC01LS, which has the LS mutation that increases the half-life of the antibody from 14 to 71 days, can protect infants born to HIV-infected mothers from contracting the virus during breastfeeding (the IMPAACT or P1112 trial).
But even before the AMP study results are in, researchers are already preparing for the possibility that it will take more than one bNAb for optimal protection against HIV infection, just as it does for therapy. “When it comes to getting a good clinical agent, we’re going to need a combination,” says Bette Korber, a computational biologist at Los Alamos National Laboratory.
Korber gave a somewhat sobering presentation at HIVR4P that suggested it may even take as many as four bNAbs for a prophylactic combination that would be effective globally. This is because the potency and breadth of the different bNAbs in development are variable against different clades of the virus. Even though all of these antibodies neutralize broadly enough to earn the distinction of being bNAbs, many of the individual antibodies are completely ineffective at neutralizing a significant fraction of viruses in laboratory panels. For example, the antibodies VRC01 and 3BNC117 don’t neutralize clade C viruses well, while PGT121 poorly neutralizes viruses from clade A as well as the CRF01 and CRF02 recombinants. Therefore the more antibodies that are combined, the harder it will be for viral escape to occur.
One antibody that seems to neutralize almost all viruses in laboratory panels, including those that others don’t, is 10E8. This antibody targets the gp41 membrane-proximal external region of the virus, which is highly conserved across clades. Unfortunately, there are now safety concerns with this antibody, calling its utility in prophylaxis into question. Koup reported that investigators observed large patches of erythema or redness at the injection sites in seven of eight individuals who received this antibody. Biopsies showed evidence of panniculitis, a group of diseases that result in the inflammation of fat tissues under the skin, and lymphocytic inflammation. No adverse event or safety concerns have arisen with VRC01 in the AMP studies. “This antibody is acting differently than other antibodies,” says Koup, who went as far as to cross 10E8 off the list of potential antibodies for HIV prophylaxis. “Until we can figure out what’s going on here, 10E8 should be off the table.”
A tri-specific antibody engineered by scientists at Sanofi and the VRC that combines the antigen-binding fragment or Fab of three bNAbs, including 10E8, into one molecule is slated to enter clinical trials. But Koup warns that the safety issues with 10E8 must be addressed before the tri-specific antibody enters clinical trials. However, researchers present at HIVR4P speculated that there may be less concern with 10E8 as part of the tri-specific, given it only contains the binding region and not the entire antibody.
In addition to the tri-specific, many groups are working to optimize bNAbs to be more potent and have longer half-lifes, as well as to test combinations of antibodies that would have a greater likelihood of protecting against HIV. The longer-acting antibodies 3BNC117-LS and 10-1074-LS, developed by scientists at Rockefeller University, are already being tested alone and in combination in a Phase I trial involving both HIV-infected and uninfected volunteers. But Korber tempered expectations that just two antibodies will be enough. “We can’t lose heart if two antibodies fail. It doesn’t mean we’re not going to succeed,” she says.
Her words could serve as a metaphor for the HIV vaccine field more broadly. Despite many failed attempts at inducing protective immunity, researchers seem more optimistic than ever that scientific advances will ultimately lead to success. While the path to developing effective vaccines and antibodies against HIV may still be long, it is at least becoming clearer.