November 22, 2019
Proven against Ebola, a vector shows its broader potential
Regulatory approval of a vesicular stomatitis virus-based vaccine paves the way for use of this viral vector in other vaccines.
Michael Dumiak
In November, the European Union approved the first vaccine against the deadly Ebola virus. Having an effective vaccine that can be deployed whenever outbreaks occur—including the one that is still simmering in the Democratic Republic of Congo that has so far claimed the lives of 2,200 people—is a milestone.
That vaccine, now referred to as Ervebo, was developed at breakneck speed during the devastating Ebola outbreak in West Africa in 2014. The science that gave rise to it, though, was 20 years in the making. This science, and the stamp of regulatory approval, may open the door to other vaccine advances.
The candidate that would become Ervebo was first developed in the early 2000s by researchers at the Public Health Agency of Canada (PHAC). But it remained dormant there for many years: clinical data was very difficult to acquire and, until more recently, the effort drew no steady or adequate resources to support it.
It was the crisis sparked by the largest-yet outbreak of Ebola virus disease in 2014 that marshalled momentum decisive enough to bring the novel vaccine forward. By the time Ebola broke out that year in Guinea, Liberia, and Sierra Leone, the vaccine candidate had already been licensed from PHAC to a subsidiary of the biotech NewLink Genetics. With the West African outbreak growing dire, NewLink began a Phase I study of the vaccine candidate, forming a steering committee on development involving PHAC, the World Health Organization, and the U.S. National Institutes of Health (NIH). In November 2014, NewLink then licensed rights to the candidate to Merck. From there, development raced into high gear during the rest of the outbreak and through its subsiding in 2016.
The vaccine was deployed during the ongoing Ebola outbreak in Congo—now the second-largest in history—under compassionate use protocols, and delivered to thousands of people. A preliminary analysis of data from the field estimates its efficacy at 97.5%; a more detailed analysis is being prepared for peer-reviewed publication. Approval of the Ebola vaccine not only offers new hope for future outbreaks, it also lends researchers confidence for further development of vaccines using the vector in Ervebo.
Ervebo is built on the back of the vesicular stomatitis virus (VSV). VSV is sometimes called Indiana vesiculovirus or vesicular stomatitis Indiana virus. Gary Kobinger, director of the Research Center on Infectious Diseases at the Université Laval in Montreal and a key figure in Ebola research, is one of many who thinks VSV may have potential as a successful vector for other vaccines beyond Ebola. Ervebo’s approval may just be the start. “It’s going to increase confidence at all levels, not just from the scientific community, but more importantly, from the public and from regulatory agencies,” Kobinger says.
More than 270,000 people in Africa have received Ervebo in the last five years as part of clinical trials or under emergency compassionate use protocols. This provides a significant amount of safety data. While every new vaccine application will need to be evaluated in the same ways, Kobinger says working on a licensed vaccine platform may ease that task.
“This is going to be very useful in the future. VSV would be one of a handful of new platforms in the last 20 years,” he says. “These are exciting days for vaccinologists to see if the VSV platform can be used and applied to protect against other pathogens.” Researchers are already employing VSV as a vector in vaccine research for other hemorrhagic fevers such as Lassa and Marburg, as well as against influenza, tuberculosis (TB), and HIV.
“While the Congo outbreak continues in a tragic way, it would have been far worse without deployment of Merck’s Ebola vaccine,” says Mark Feinberg, IAVI’s president and chief executive officer who, while at Merck, helped lead the collaborative effort that expedited Ebola vaccine development. “It’s a tremendous public health accomplishment. But it’s not only that there is a licensed Ebola vaccine that has a strong record of efficacy and tolerability, and the ability to be implemented. The fact that the vaccine was licensed by the European Medicines Agency, and will hopefully soon be licensed by the FDA [U.S. Food and Drug Administration], is a very positive precedent that makes it much more feasible to imagine developing additional vaccines based on the VSV platform.”
VSV is an RNA virus in the rhabdovirus family that affects, among other animals, cows, pigs, and horses. Natural infection causes vesicular lesions of the tongue, teats, and hooves of livestock, a mild infection clearing within two weeks. VSV can infect humans but does so only rarely, causing mild flu-like symptoms, and is mostly inapparent, says Chris Parks, executive director of IAVI’s Vaccine Design and Development Laboratory. His team is using VSV as a vector in developing experimental vaccine candidates against HIV, Lassa, and Marburg.
Yale’s John Rose and his colleagues developed an experimental HIV vaccine using VSV back in 2001 and have worked with the virus for many years (Cell 106, 539, 2001). Rose’s work is a wellspring for research on VSV as a vector—including the work at PHAC that would eventually lead to the Ebola vaccine.
A vaccine vector is used to transport genes or proteins from another virus to trigger an immune response. The vector is the delivery system, and can be a live virus, an attenuated or weakened virus, or an inactivated or killed virus. There are very few licensed human vaccines that utilize viral vectors; among them are those for Japanese encephalitis and dengue. Both of these vaccines are based on a yellow fever viral vector. Now there is also Ervebo.
There are several features that make VSV an attractive vaccine vector. Its relatively small genome consists of a single molecule of RNA encoding for only five major proteins (see Figure), one of which, the glycoprotein (G), mediates its attachment to the host cell receptor, allowing it to enter the cell and hijack it in order to replicate and spread. The virus’s small genome makes it easy to manipulate, says Andrea Marzi, a virologist and Ebola researcher at the National Institute of Allergy and Infectious Diseases’ Rocky Mountain Laboratories in Hamilton, Montana. Cytomegalovirus, by comparison, another virus vector used in experimental vaccine development, has close to 200 genes, making it much more complicated to manipulate.
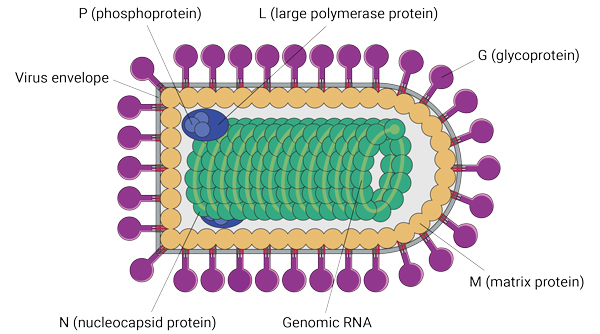 A bullet-shaped virus could hit the target for many future vaccines. Vesicular stomatitis virus, or VSV, is made up of five proteins, its viral envelope, and genomic RNA.
A bullet-shaped virus could hit the target for many future vaccines. Vesicular stomatitis virus, or VSV, is made up of five proteins, its viral envelope, and genomic RNA.
As a vector, VSV offers other advantages. It is a replicating virus, but its low prevalence in humans means there is no pre-existing immunity to VSV in the general population that could potentially limit its efficacy. Because VSV is replication-competent, Kobinger says, it is also a strong candidate to induce lasting responses to the antigen it is carrying. But because its RNA does not integrate into a host cell, there’s less risk for oncogenesis or mutagenesis.
How VSV works its magic in stimulating a powerful immune response is still an open question, Kobinger says. “We don’t exactly know all the details. Because it was used first for an Ebola vaccine, there was a very small community working on it. We don’t know exactly the specific molecule or determinants that are involved in the immune stimulation,” he says.
Lab work with VSV goes back a long time. Parks has seen papers from the 1930s detailing VSV research. But for decades it seemed like VSV might never be a vehicle for vaccines due to its potential neurotoxicity.
These concerns were what caused Kobinger to drop the virus for a time, figuring it was very unlikely that a vaccine using VSV would ever go into advanced clinical use. Injected directly into the brain of mice or other animals, VSV can cause neurotoxicity, Parks says. Researchers now know that the neurotoxicity caused by the wild-type virus probably has something to do with the G protein, which allows the virus to replicate extensively in the brain. That question is still not fully answered scientifically, but, as borne out by Ervebo’s safety data, vaccine researchers eventually discovered how to neutralize it.
Starting with the non-clinical Yale work in 1999 and 2001 of Eli Boritz, Rose, and others in labs at Tulane, Duke, the University of California, San Francisco, and at Rockefeller University in New York, and then, finally, with a team including Michael Garbutt of PHAC’s National Microbiology Laboratory and Heinz Feldmann (who supervised Marzi’s postdoc there before both moved to Montana), researchers plotted a way around the neurotoxicity issue. They did it by knocking out the G protein in VSV to make chimeras (J. Virol. 78(10), 5458, 2004). The reputation that VSV had because of the toxicity in the brains of macaques—the virulence factor, Kobinger calls it—was defanged.
“If you remove the G protein, you really don’t see those neurotoxicity effects,” he says. This was quickly reflected in lab experiments done using VSV. Feldmann’s group created VSV chimeras using glycoproteins from Lassa, Ebola, and Marburg viruses. Feldmann was once special pathogen chief at the PHAC’s National Microbiology Laboratory (where he was succeeded by Kobinger). Along with Ute Ströher and other PHAC researchers, the group drew upon the previous work of Rose and others and knocked out the VSV G protein, replacing it with the Ebola virus glycoprotein (CMAJ 189, E1326, 2017). The VSV-Ebola chimera Feldmann and his colleagues created is the origin of the vaccine developed by Merck.
Merck compiled safety and efficacy data for Ervebo from eight Phase I clinical trials and five Phase II and Phase III studies in a variety of countries and populations including Liberia, Sierra Leone, Guinea, Canada, Spain, and the U.S. These trials involved 15,996 people, including 234 children, 536 elderly people, 261 pregnant women, and 22 HIV-infected volunteers, showing limited local reactions—pain and swelling, for the most part—of mild to moderate severity.
Now an Ebola vaccine is on hand with perhaps others on the way (see When Ebola returns, will the world be ready?, IAVI Report, Vol. 19, No. 4, 2015). Marzi says the lessons for vaccine development are clear: they show the need for basic research and ongoing vaccine design and development in advance of an epidemic and the need for vaccine platforms that could be swiftly applied to multiple pathogens. (Ann. Rev. Microbiology 72, 423, 2018) Researchers hope this is what’s in the cards for VSV.
There are many groups investigating VSV as a vector. Kobinger’s lab in Montreal, the team Parks is leading at IAVI, the University of Manitoba, University of Texas, NIH’s Rocky Mountain Laboratories, and the Medical University of Innsbruck are all part of collaborative efforts developing candidates against HIV, Ebola, and hemorrhagic fevers. Yale is experimenting with a VSV-based candidate against severe acute respiratory syndrome virus (SARS); the State Key Laboratory of Veterinary Biotechnology is experimenting with a candidate against Middle East respiratory syndrome coronavirus (MERS); and the University of Miami, which, along with Rocky Mountain labs, is pursuing candidates against Zika. The vector is also linked to candidates against flu, TB, and even plague (J. Virol. doi:10.1128/JVI.05991-11, and see survey Hum. Vaccin. Immunother. doi:10.1080/21645515.2019.1649532).
VSV may even prove useful as an oncology therapy, an avenue pursued by the Medical University of Innsbruck’s Dorothee von Laer. The concept is that the virus would infect and replicate in tumor cells and lyse them, acting as a more benign alternative to chemotherapy. Von Laer’s small biotech firm ViraTherapeutics was absorbed by Boehringer Ingelheim to further develop this approach.
Janine Kimpel is a virologist, speed chess champion, and one of von Laer’s protégés in Innsbruck. She and her group are using the same VSV vehicle, developed by von Laer for tumor treatment and dubbed VSV-GP, as a vector for HIV vaccine candidates. VSV-GP is a replication-competent chimeric virus, using the backbone of VSV with its G protein exchanged for the glycoprotein of lymphocytic choriomeningitis virus (LCMV). The LCMV glycoprotein has a very broad cell tropism.
Kimpel’s team is currently preparing VSV-GP candidates for preclinical trials in Paris as part of the effort under the European HIV Alliance (EHVA, www.ehv-a.eu). EHVA is an umbrella group with 41 partners working in discovery, immune profiling, and clinical trial platforms to develop novel HIV vaccine candidates. Kimpel’s team has added an HIV Env protein to their VSV-GP vector and are hoping that they can take advantage of VSV’s efficiency in incorporating foreign glycoproteins to boost the chances of inducing good antibody responses against HIV. The difference between Kimpel’s approach and some of the other groups has to do with how Env is added to the chimeric vector, in this case adding a gene expressing HIV Env to a VSV chimera. As the LCMV glycoprotein is mediating replication in the vaccine formulation, the modified HIV Env does not need to be infectious, and the vector’s target cell range can be more broad. As the team has shown, the antibody responses are not neutralizing to the vector itself (J. Virol. 88(9), 4897, 2014).
There are other concepts at play in the HIV field. Ma Luo of the University of Manitoba is running a group experimenting with expressed conserved HIV peptides carried by modified VSV; Jonathan Fuchs at the University of California, San Francisco, in collaboration with Profectus and U.S. National Institute of Allergy and Infectious Diseases, ran a safety trial with a VSV HIV-1 gag vaccine candidate four years ago (Open Forum Infect. Dis. doi:10.1093/ofid/ofv082). Kobinger and Parks are also both pursuing novel HIV vaccine candidates and employing different strategies to do so.
The concept in Kobinger’s lab is to use an Ebola glycoprotein and show it as a target carried by the VSV vector to a sub-population of antigen-presenting cells. That vector will also carry an HIV protein. “There are spikes on top, which are normally the glycoprotein used by the viral particle for entry,” Kobinger says. “Let’s say for the sake of this that maybe 30% of the spikes are Ebola and 70% of the spikes are HIV. That 30% is enough to target the particle.” Ebola glycoprotein should bring the VSV vaccine carrier in antigen-presenting cells, he says. The plan is to bring two or three candidates using this strategy into a large preclinical study.
Parks’ lab is replacing the VSV G protein completely with HIV Env so it is the only glycoprotein expressed by the vector. As opposed to Kimpel’s approach, Parks’ team electroporates Vero-CD4-CCR5 cells with plasmid expressing VSV G protein, and then infects them with VSV with deleted G and HIV Env. The progeny virions incorporate Env. The team tested it in preclinical monkey studies, which Feinberg says showed a 70% protective effect against repeat, low-dose challenges using SHIV, the engineered virus containing both HIV genes and those of simian immunodeficiency virus (SIV). “We tried to repeat it in a second study,” Parks says, but in that case the vaccine was notably less effective. “In both studies, we saw quite strong antibody responses.” Parks and colleagues are now working to figure out why the VSV vector was efficacious in one preclinical study and not another. “We think we understand why: it’s some technical issues with the vaccine production process. We hope to do another monkey study next year to try and sort it out, and hopefully it will work again.”
The vaccine production in this case is complex. It starts with cell cultures, using lines of cells called Vero cells that were first extracted from green monkey kidney cells. Large quantities of Vero cells are grown in a manufacturing facility and these are infected with viral vector. The virus replicates under incubation, and then the virus that is released from infected cells is harvested, purified, and concentrated sufficiently for use in a vaccine. But standard Vero cells won’t work for IAVI’s HIV candidate because the cell line does not express the receptors recognized by HIV Env: they needed to modify the Vero line to fix this. Experiments using the first modified line were promising, but only a short-term solution, as the desired receptors gradually switched off expression. A second-generation line provided higher immune response, but efficacy was diminished. The team’s now experimenting with modifying the process to resolve this discrepancy.
IAVI is working with Batavia Biosciences of Leiden, the Netherlands, as a manufacturing partner, Feinberg says. “We’re applying innovative manufacturing technologies that will hopefully be scalable and flexible, and make vaccine manufacturing—especially for outbreak-related pathogens—more simple and effective.” Feinberg doesn’t think viral vector-based vaccines are necessarily more complicated to manufacture than other vaccines.
Other pathogens are also under the microscope as potential targets for VSV candidates. IAVI is pursuing candidates against Lassa fever, supported by the Coalition for Epidemic Preparedness Innovations (CEPI), and Marburg. IAVI’s VSV vectors for Lassa and Marburg, which were licensed from PHAC, are identical to the vector used in Ervebo. “That’s an advantage because you know how the vector performs, and to the extent that the backbone can be one of the factors that influences your ability to produce and deliver it, that’s a benefit,” says Feinberg. “That’s not the entirety of the vaccine, but at least these important elements of the vaccine are ones for which you will have set a precedent by licensure.”
In 2018 CEPI also issued a grant to Profectus (which is currently in process of selling some of its research and development assets to Aurobindo Pharmaceuticals) and Emergent Biosciences to work on a Lassa vaccine based on its VesiculoVax vaccine delivery platform, which is also derived from VSV.
Feinberg says VSV’s profile is well suited for outbreak-associated pathogens like Ebola, Lassa fever, and Marburg. “Others, though, are applying it for additional infectious diseases like Nipah virus or chikungunya. This demonstrates the tremendous flexibility of the VSV platform.”
All told, it is the vector’s viral structure that makes it flexible, says Trina Racine, a virologist, biotech consultant, and former colleague in Kobinger’s lab. “We can pack in quite large antigens, and you can potentially express multiple antigens. In an ideal world, maybe one day we can produce a vaccine that is for Ebola and HIV at the same time.” Marzi’s group has experimented with a Zika-Ebola antigen in mice.
Researchers are also working with another VSV strain, VSV New Jersey, and other related vesiculoviruses like Alagoas, Maraba, and Chandipura. The idea of using alternate strains is to avoid potential effects of anti-vector immunity, especially if VSV Indiana becomes more widely used for vaccination in humans.
It also takes more than a vector to get vaccines to people. As the latest deadly outbreak illustrates, delivering Ervebo and other still-experimental Ebola vaccines into remote, conflict-stricken, and inaccessible areas while kept at -76 to -112 degrees Fahrenheit remains a challenge.
When the outbreak hit Western Africa five years ago, Marzi went to Liberia to help the U.S. Centers for Disease Control and Prevention and Médecins Sans Frontières build a diagnostic lab and Ebola treatment unit in Monrovia. She saw the epidemic firsthand. “We have a great vaccine now, but people have to keep in mind what it was developed for,” she says. “It is really developed to be used in an emergency situation, like an outbreak. For Ebola virus, we never thought it feasible until there was an epidemic that we would have to vaccinate the population of an entire country, which might now become necessary, particularly in Congo.”
Marzi and her group just published results from a new set of dosing experiments, aiming to see if Ebola vaccine can eventually be stretched further by delivering it in lower, but still effective amounts (EBioMedicine 49, 223, 2019). As a vaccine vector, though, VSV may end up stretching a lot further than that.
Michael Dumiak, based in Berlin, reports on global science, public health, and technology.